GATK4 Germline SNPs Indels#
22/03/2022
The GATK Germline SNPs Indels is a bioinformatic pipeline widely used for variant calling. It takes BAM as an input and generates a VCF file as an output. As WDL is developed and highly supported by the Broad Institute, this pipeline has been standardized by WDL and open sourced on GitHub ( ).
).
In this case study, we modify and demonstrate the performance of the GATK Germline SNPs Indels pipeline on the SeqsLab platform with the GATK Processing for Variant Discovery pipeline, gatk4-data-processing (), which preprocesses a FASTQ data to a BAM so that the whole WDL pipeline can start with a FASTQ file.
Main WDL#
For this demo, we created a main WDL which includes the two WDL subworkflows below. Both processing-for-variant-discovery-gatk4.wdl and haplotypecaller-gvcf-gatk4.wdl files are copied from the GATK Github with some modifications. The main WDL file, the modified haplotypecaller-gvcf-gatk4.wdl (), and the modified processing-for-variant-discovery-gatk4.wdl () can be found in the Atgenomix Github ().
version 1.0
import "../../gatk4-data-processing/processing-for-variant-discovery-gatk4.wdl" as mapper
import "../../gatk4-germline-snps-indels/haplotypecaller-gvcf-gatk4.wdl" as caller
# WORKFLOW DEFINITION
workflow e2e_HaplotypeCallerGvcf_GATK4 {
input {
String sample_name
String ref_name
Array[File] fastq_files
File ref_fasta
File ref_fasta_index
File ref_dict
File? ref_alt
File ref_sa
File ref_ann
File ref_bwt
File ref_pac
File ref_amb
File dbSNP_vcf
File dbSNP_vcf_index
Array[File] known_indels_sites_VCFs
Array[File] known_indels_sites_indices
String gatk_path
String gotc_path
String bwa_commandline = "bwa mem -K 100000000 -p -v 3 -t 16 -Y $bash_ref_fasta"
Int compression_level = 5
String gatk_docker = "us.gcr.io/broad-gatk/gatk:4.2.0.0"
String gotc_docker = "us.gcr.io/broad-gotc-prod/genomes-in-the-cloud:2.4.7-1603303710"
String python_docker = "python:2.7"
Int flowcell_small_disk = 100
Int flowcell_medium_disk = 200
Int agg_small_disk = 200
Int agg_medium_disk = 300
Int agg_large_disk = 400
Int preemptible_tries = 3
Boolean make_gvcf = true
Boolean make_bamout = false
String samtools_path = "samtools"
}
call mapper.PreProcessingForVariantDiscovery_GATK4 {
input:
sample_name = sample_name,
ref_name = ref_name,
fastq_files = fastq_files,
ref_fasta = ref_fasta,
ref_fasta_index = ref_fasta_index,
ref_dict = ref_dict,
ref_alt = ref_alt,
ref_sa = ref_sa,
ref_ann = ref_ann,
ref_bwt = ref_bwt,
ref_pac = ref_pac,
ref_amb = ref_amb,
dbSNP_vcf = dbSNP_vcf,
dbSNP_vcf_index = dbSNP_vcf_index,
known_indels_sites_VCFs = known_indels_sites_VCFs,
known_indels_sites_indices = known_indels_sites_indices,
bwa_commandline = "bwa mem -K 100000000 -p -v 3 -t 16 -Y $bash_ref_fasta",
compression_level = 5,
gatk_docker = "us.gcr.io/broad-gatk/gatk:4.2.0.0",
gatk_path = gatk_path,
gotc_docker = "us.gcr.io/broad-gotc-prod/genomes-in-the-cloud:2.4.7-1603303710",
gotc_path = gotc_path,
python_docker = "python:2.7",
flowcell_small_disk = 100,
flowcell_medium_disk = 200,
agg_small_disk = 200,
agg_medium_disk = 300,
agg_large_disk = 400,
preemptible_tries = 3
}
call caller.HaplotypeCallerGvcf_GATK4 {
input:
input_bam = PreProcessingForVariantDiscovery_GATK4.analysis_ready_bam,
ref_dict = ref_dict,
ref_fasta = ref_fasta,
ref_fasta_index = ref_fasta_index,
make_gvcf = true,
make_bamout = false,
gatk_docker = "us.gcr.io/broad-gatk/gatk:4.2.0.0",
gatk_path = gatk_path,
gitc_docker = "us.gcr.io/broad-gotc-prod/genomes-in-the-cloud:2.4.7-1603303710",
samtools_path = "samtools"
}
output {
File e2e_analysis_ready_bam = PreProcessingForVariantDiscovery_GATK4.analysis_ready_bam
File e2e_output_vcf = HaplotypeCallerGvcf_GATK4.output_vcf
File e2e_bqsr_report = PreProcessingForVariantDiscovery_GATK4.bqsr_report
File e2e_output_vcf_index = HaplotypeCallerGvcf_GATK4.output_vcf_index
File e2e_duplication_metrics = PreProcessingForVariantDiscovery_GATK4.duplication_metrics
}
}
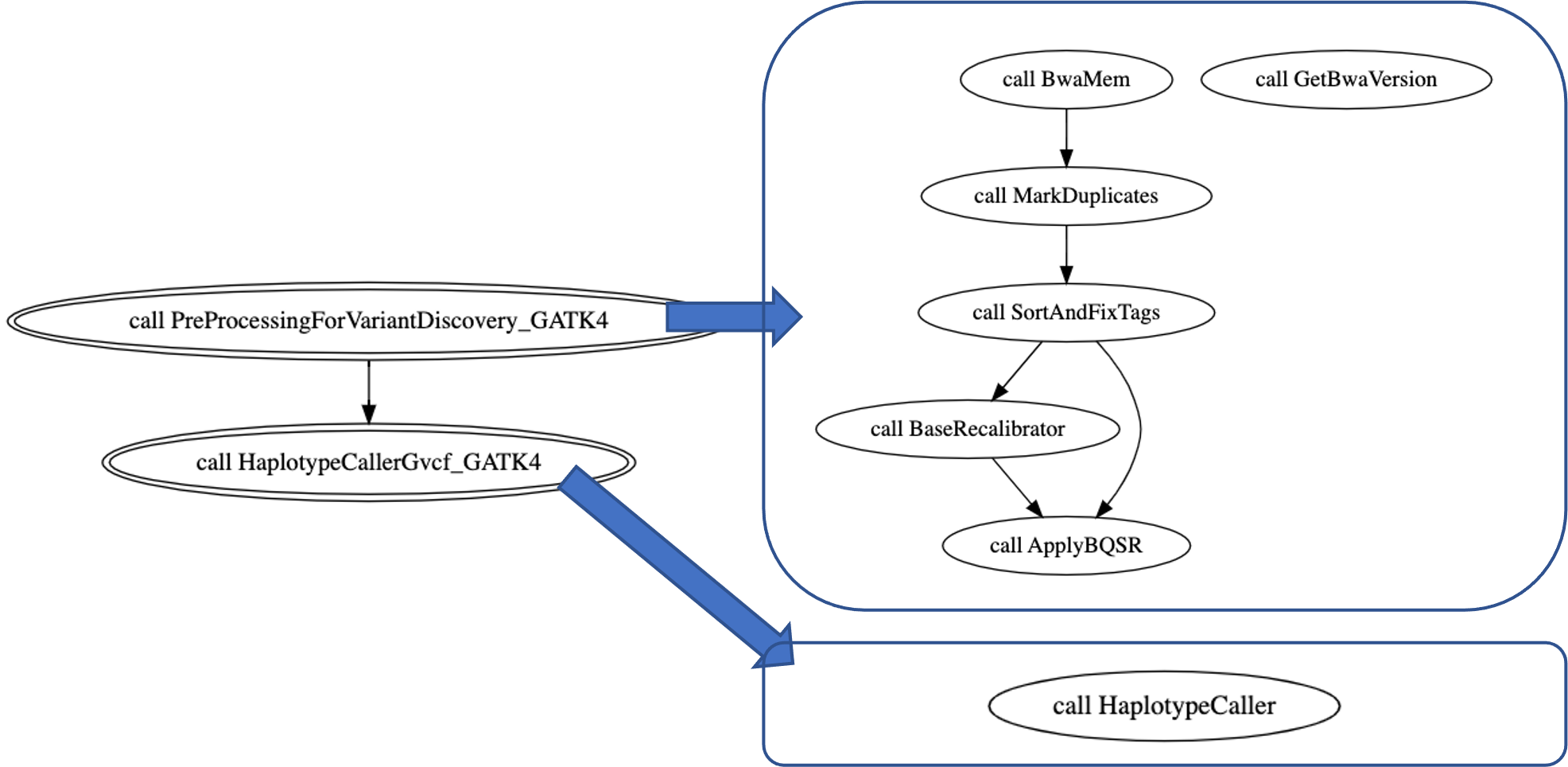
WOM graphs#
Based on the Cromwell WOMtool, we will get the following digraphs to describe this pipeline. The main WDL includes two subworkflows: PreProcessingForVariantDiscovery_GATK4 and HaplotypeCallerGvcf_GATK4. The former is composed of the following tasks: BwaMem, GetBwaVersion, MarkDuplicateds, SortAndFixTags, BaseRecalibrator, and ApplyBQSR. The latter contains only one task: HaplotypeCaller.
CLI commands#
Once we have the WDL files, we also need to have the inputs.json () file shown below and a Docker image for packaging the runtime environment.
{
"e2e_HaplotypeCallerGvcf_GATK4.ref_sa": "hg38/Homo_sapiens_assembly38.fasta.64.sa",
"e2e_HaplotypeCallerGvcf_GATK4.ref_amb": "hg38/Homo_sapiens_assembly38.fasta.64.amb",
"e2e_HaplotypeCallerGvcf_GATK4.ref_ann": "hg38/Homo_sapiens_assembly38.fasta.64.ann",
"e2e_HaplotypeCallerGvcf_GATK4.ref_bwt": "hg38/Homo_sapiens_assembly38.fasta.64.bwt",
"e2e_HaplotypeCallerGvcf_GATK4.ref_pac": "hg38/Homo_sapiens_assembly38.fasta.64.pac",
"e2e_HaplotypeCallerGvcf_GATK4.ref_dict": "hg38/Homo_sapiens_assembly38.dict",
"e2e_HaplotypeCallerGvcf_GATK4.ref_name": "hg38",
"e2e_HaplotypeCallerGvcf_GATK4.dbSNP_vcf": "hg38/Homo_sapiens_assembly38.dbsnp138.vcf",
"e2e_HaplotypeCallerGvcf_GATK4.gatk_path": "/gatk/gatk-4.2.0.0/gatk",
"e2e_HaplotypeCallerGvcf_GATK4.gotc_path": "/usr/bin/",
"e2e_HaplotypeCallerGvcf_GATK4.ref_fasta": "hg38/Homo_sapiens_assembly38.fasta",
"e2e_HaplotypeCallerGvcf_GATK4.fastq_files": [
"r1.fq",
"r2.fq"
],
"e2e_HaplotypeCallerGvcf_GATK4.sample_name": "NA12878",
"e2e_HaplotypeCallerGvcf_GATK4.dbSNP_vcf_index": "hg38/Homo_sapiens_assembly38.dbsnp138.vcf.idx",
"e2e_HaplotypeCallerGvcf_GATK4.ref_fasta_index": "hg38/Homo_sapiens_assembly38.fasta.fai",
"e2e_HaplotypeCallerGvcf_GATK4.known_indels_sites_VCFs": [
"hg38/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz",
"hg38/Homo_sapiens_assembly38.known_indels.vcf.gz"
],
"e2e_HaplotypeCallerGvcf_GATK4.known_indels_sites_indices": [
"hg38/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz.tbi",
"hg38/Homo_sapiens_assembly38.known_indels.vcf.gz.tbi"
]
}
Create request.json#
We used the following command to create the request.json file:
jobs request workspace=seqslabwu2 working-dir="./seqslab-workflows/gatk-best-practice" run-name="Demo-1" workflow-url="https://dev-api.seqslab.net/trs/v2/tools/trs_3hw1vuGqCyMEWqY/versions/1.0.8/WDL/files/"
Run Wes Job#
We used the following command to run the job:
jobs run workspace=seqslabwu2 working-dir="./seqslab-workflows/gatk-best-practice"
Parallelization Settings#
Default Non Parallel Settings#
On the SeqsLab platform, you can benefit from implicit data parallelization by changing the operator pipeline settings inside the workflow_params section in the request.json file instead of modifying any WDL pipeline. By default, when you generate your request.json file, the operator pipeline is configured as shown in the following example.
{
"fqn": "e2e_HaplotypeCallerGvcf_GATK4.PreProcessingForVariantDiscovery_GATK4.SortAndFixTags.ref_fasta_index",
"operators": {
"format": {
"class": "com.atgenomix.seqslab.piper.operators.format.SingularFormat"
},
"p_pipe": {
"class": "com.atgenomix.seqslab.piper.operators.pipe.PPipe"
}
},
"pipelines": {
"call": [
"format",
"p_pipe"
]
}
}
This setting describes how FQN, which might be a File, Array[File] or Array[Array[File]], is going to be processed. With the above settings, SingularFormat and PPipe, ref_fasta_index file will be loaded as a single file and rendered into the provided command as other input variables.
Note
Users can only configure operator pipelines on input FQN at task level. SeqsLab will support data manipulation at output FQN in the future.
Parallel Settings#
In order to benefit from parallel computation, you need to know where to apply different operator pipeline settings for splitting and merging data. For example, in this tutorial, we have FASTQ R1, R2 data recognized as FQNs, e2e_HaplotypeCallerGvcf_GATK4.PreProcessingForVariantDiscovery_GATK4.BwaMem.inFileFastqR1 and e2e_HaplotypeCallerGvcf_GATK4.PreProcessingForVariantDiscovery_GATK4.BwaMem.inFileFastqR2 in a BwaMem task. We configure a chunk operation where each FASTQ chunk will contain 3145728 reads. One reason for chunking with this number is to guarantee that each FASTQ will be around 256MB, which is a good size for an Hdfs/Hadoop operation.
{
"fqn": "e2e_HaplotypeCallerGvcf_GATK4.PreProcessingForVariantDiscovery_GATK4.BwaMem.inFileFastqR1",
"operators": {
"format": {
"auth": "",
"class": "com.atgenomix.seqslab.piper.operators.format.FastqFormat"
},
"p_pipe": {
"class": "com.atgenomix.seqslab.piper.operators.pipe.PPipe"
},
"source": {
"auth": "",
"class": "com.atgenomix.seqslab.piper.operators.source.FastqSource",
"codec": "org.seqdoop.hadoop_bam.util.BGZFCodec"
},
"partition": {
"class": "com.atgenomix.seqslab.piper.operators.partition.FastqPartition",
"parallelism": "3145728"
}
},
"pipelines": {
"call": [
"format",
"p_pipe"
],
"input": [
"source",
"partition",
"format"
]
}
}
Before the next task, MarkDuplicates, we need BAM repartition after BwaMem. As such, we need to preprocess the input of MarkDuplicates before running the command. We add an operator pipeline setting for FQN, input_bam inside MarkDuplicates task. Through this setting, input_bam will be loaded as BamSource, partitioned into 155 parts and written into another Bam file. The expected result is the command of MarkDuplicates.
{
"fqn": "e2e_HaplotypeCallerGvcf_GATK4.PreProcessingForVariantDiscovery_GATK4.MarkDuplicates.input_bam",
"operators": {
"format": {
"auth": "",
"class": "org.bdgenomics.adam.cli.piper.BamFormat"
},
"p_pipe": {
"class": "com.atgenomix.seqslab.piper.operators.pipe.PPipe"
},
"source": {
"auth": "",
"class": "org.bdgenomics.adam.cli.piper.BamSource"
},
"partition": {
"ref": "abfss://static@seqslabbundles.dfs.core.windows.net/reference/38/GRCH/ref.dict",
"class": "org.bdgenomics.adam.cli.piper.BamPartition",
"parallelism": "abfss://static@seqslabbundles.dfs.core.windows.net/system/bed/38/contiguous_unmasked_regions_155_parts"
}
},
"pipelines": {
"call": [
"format",
"p_pipe"
],
"input": [
"source",
"partition",
"format"
]
}
}
Unlike a non-parallel case, data are partitioned in a parallel pipeline. We cannot further use SingularFormat to load those intermediate files. Instead, PartitionedFormat is introduced to read files which are already partitioned. For example, after MarkDuplicates data are partitioned into 155 regions for input_bam of SortAndFixTags task, PartitionedFormat needs to be provided as a loading method.
{
"fqn": "e2e_HaplotypeCallerGvcf_GATK4.PreProcessingForVariantDiscovery_GATK4.SortAndFixTags.input_bam",
"operators": {
"format": {
"auth": "",
"class": "com.atgenomix.seqslab.piper.operators.format.PartitionedFormat"
},
"p_pipe": {
"class": "com.atgenomix.seqslab.piper.operators.pipe.PPipe"
}
},
"pipelines": {
"call": [
"format",
"p_pipe"
]
}
}
For other input data, such as the reference data which is not partitioned as FASTQ, BAM, and VCF the SingularFormat is no longer usable in a parallel case. Instead, as these reference data are shared between calculation of all data partititons, SeqsLab supports loading them with SharedFile and SharedDir format. Using the SharedFile format, it is not necessary to download a file multiple times in the same nodes.
{
"fqn": "e2e_HaplotypeCallerGvcf_GATK4.PreProcessingForVariantDiscovery_GATK4.SortAndFixTags.ref_fasta_index",
"operators": {
"format": {
"auth": "",
"class": "com.atgenomix.seqslab.piper.operators.format.SharedFile"
},
"p_pipe": {
"class": "com.atgenomix.seqslab.piper.operators.pipe.PPipe"
}
},
"pipelines": {
"call": [
"format",
"p_pipe"
]
}
}
Performance evaluation#
The following table shows the performance statistics compared between parallel and non-parallel settings. In this demonstration, we repartition the BAM file into 155 parts.
Performance (Time) |
Non-Parallel Azure D14_v2 x 1 |
Non-Parallel Azure D15_v2 x 1 |
Parallel Azure D14_v2 x 10 |
Parallel Azure D14_v2 x 20 |
|---|---|---|---|---|
GetBwaVersion |
8m:17s |
7m:59s |
8m:01s |
8m:45s |
BwaMem |
2h:32m:45s |
2h:29m:41s |
39m:20s |
32m:53s |
MarkDuplicates |
1:39m:3s |
1h:39m:49s |
22m:55s |
22m:50s |
SortAndFixTags |
1h:16m:49s |
1h:15m:54s |
7m:11s |
5m:26s |
BaseRecalibrator |
2h:7m:19s |
1h:41m:52s |
6m:39s |
6m:29s |
ApplyBQSR |
2h:1m:54s |
1h:37m:42s |
5m:22s |
4m:41s |
HaplotypeCaller |
18h:29m:19s |
15h:59m:52s |
2h:14m:55s |
1h:15m:49s |
Total |
28h:8m:14s |
24h:44m:14s |
3h:39m:02s |
2h:30m:53s |